Welcome to DataLab!
Looking for passionate Ph. D. and M.S. students who want to study the research area of data science, computational biology and bioinformatics.
If interested, please contact to Dr. Gangman Yi.
News
- [2026.01]
- Paper accepted to IEEE Access: “A Method for Consensus Anchor Graph Learning for Incomplete Multi-View Clustering"
- [2025.10]
- 김동연 박사과정이 '한국이공학진흥원(IPESK): 차세대공학연구자-최우수상'을 수상하였습니다.
- [2025.08]
- 김민섭 석사과정이 '2025년도 석사과정생연구장려금지원사업 :부정확한 레이블을 포함한 partial multi-label 데이터 환경에서의 distance metric 학습 기반 다중 레이블 분류 모델 연구' 에 선정되었습니다.
- Paper accepted to Interdisciplinary Sciences: Computational Life Sciences: “GapSense: Similarity estimation-based gap filler with TGS-reads for genome assemblies"
- Eunseo(undergraduate) joined the lab.
- [2025.07]
- 김민섭 석사과정이 'KCC 2025 우수발표논문상'을 수상하였습니다.
- 양진경 석사과정이 'KCC 2025 우수논문상'을 수상하였습니다.
- [2025.06]
- Beomsoo(undergraduate) joined the lab.
- 이승민(석사)이 '삼성전자 DX부문 신입사원 채용'에 최종 합격하였습니다.
- [2025.05]
- 김민섭 석사과정이 '서울장학재단: AI 서울테크 대학원 장학금 지원사업'의 장학생으로 선정되었습니다.
- Jaewon(undergraduate) joined the lab.
- Paper accepted to Human-centric Computing and Information Sciences: “Optimizations in Enumerating Maximal Balanced Bicliques: Pruning and Vertex Sorting”.
- [2025.03]
- 양진경 석사과정이 '2025년도 여대학원생 공학연구팀제 심화과정: 약물-표적 상호작용 예측을 위한 그래프 신경망 모델 개발 연구' 지원사업에 선정되었습니다.
- Ilkhomjon(Ph.D.), Minseop(master's), Sejin(undergraduate) joined the lab.
Research
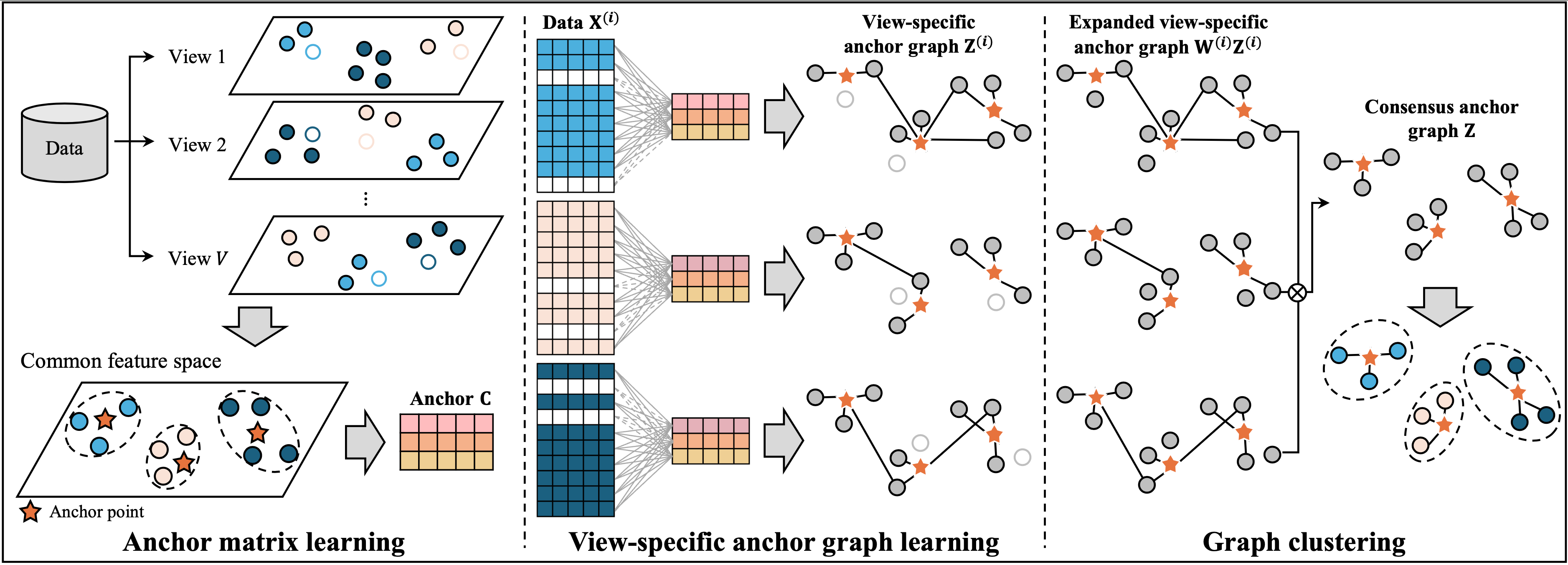
IMC
A Method for Consensus Anchor Graph Learning for Incomplete Multi-View Clustering
2026-01-25
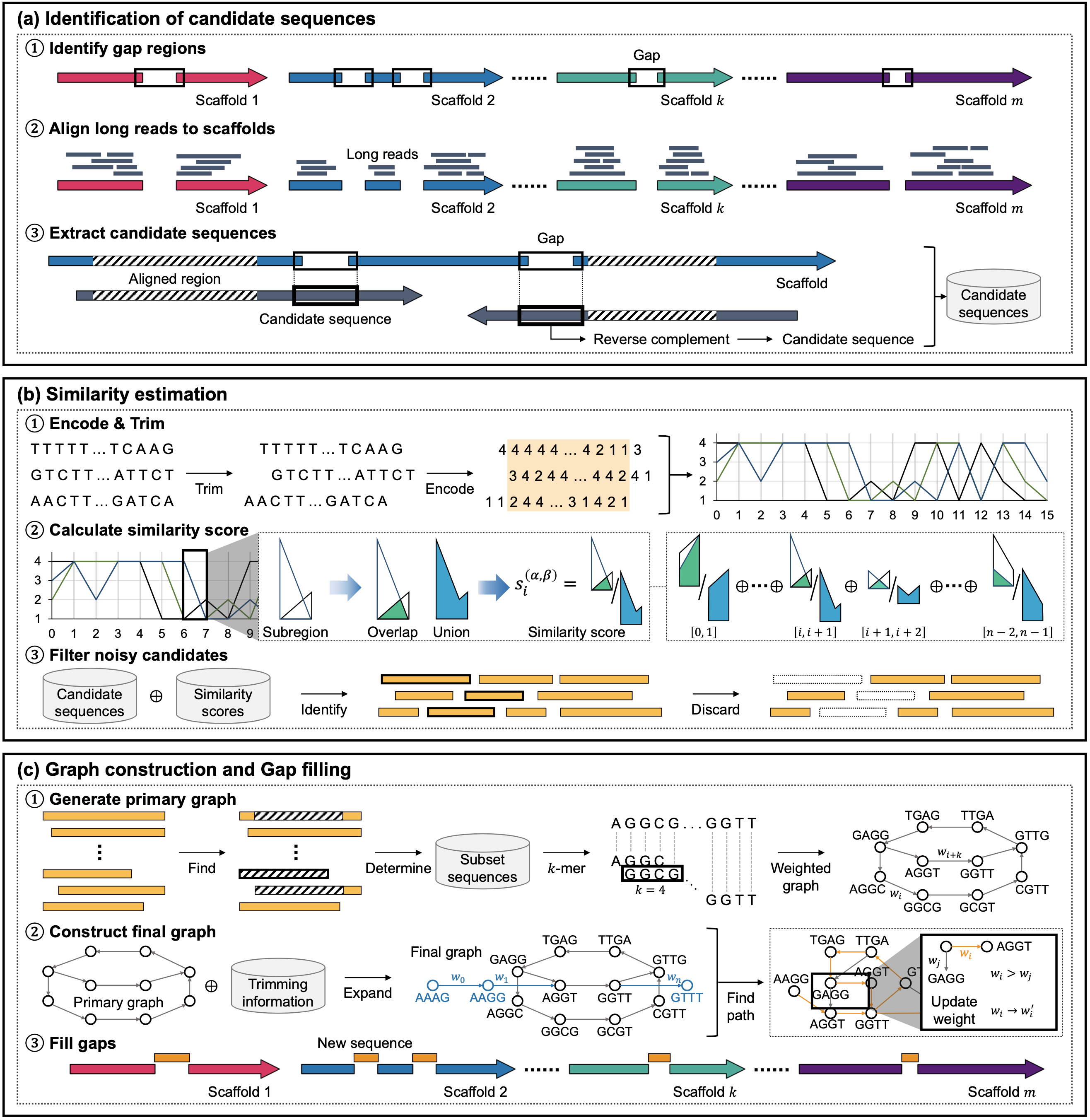
GapSense
GapSense: Similarity Estimation-Based Gap Filler with TGS-Reads for Genome Assemblies
2025-08-31
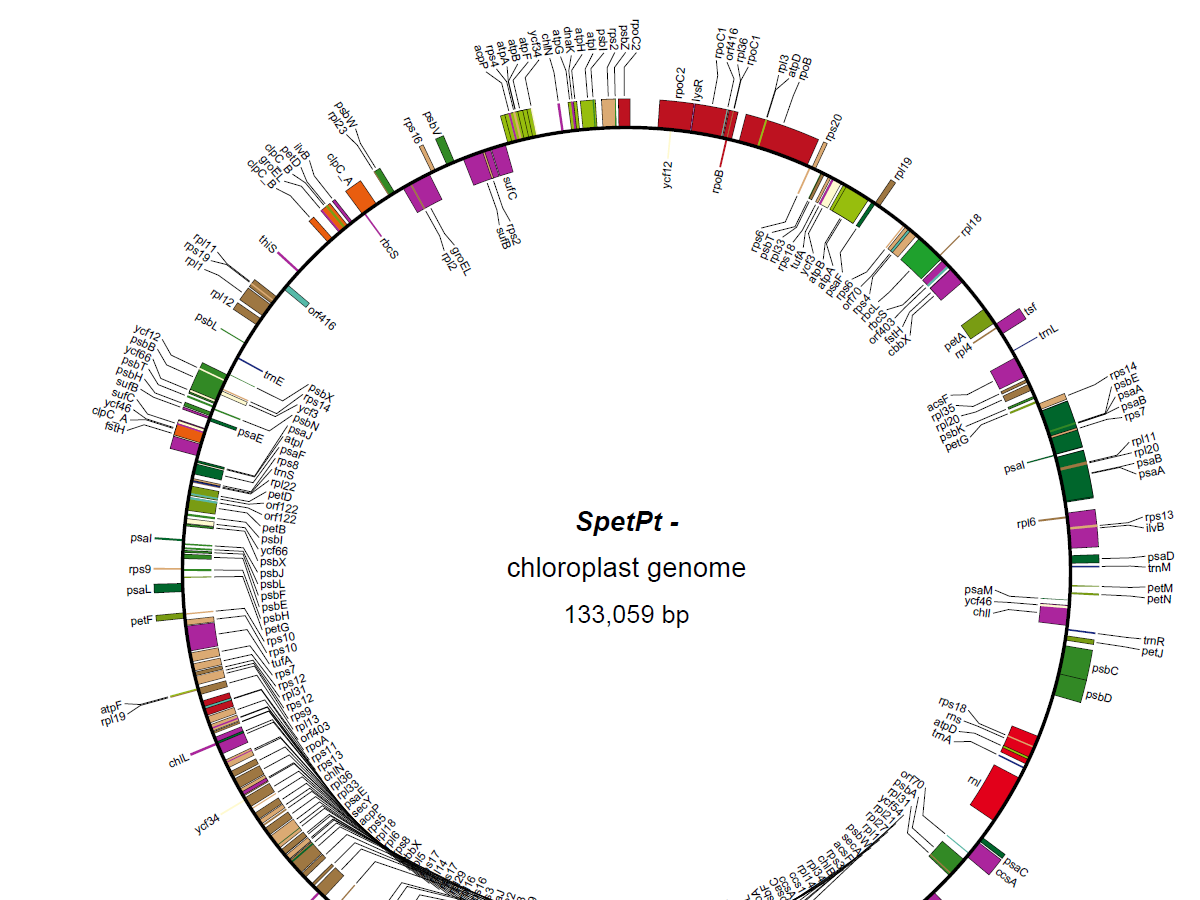
Gene Annotation
Next-generation sequencing (NGS) technologies have led to the accumulation of high-throughput sequence data from various organisms in biology. To apply gene annotation of organellar genomes for various organisms, more optimized...
2019-12-15
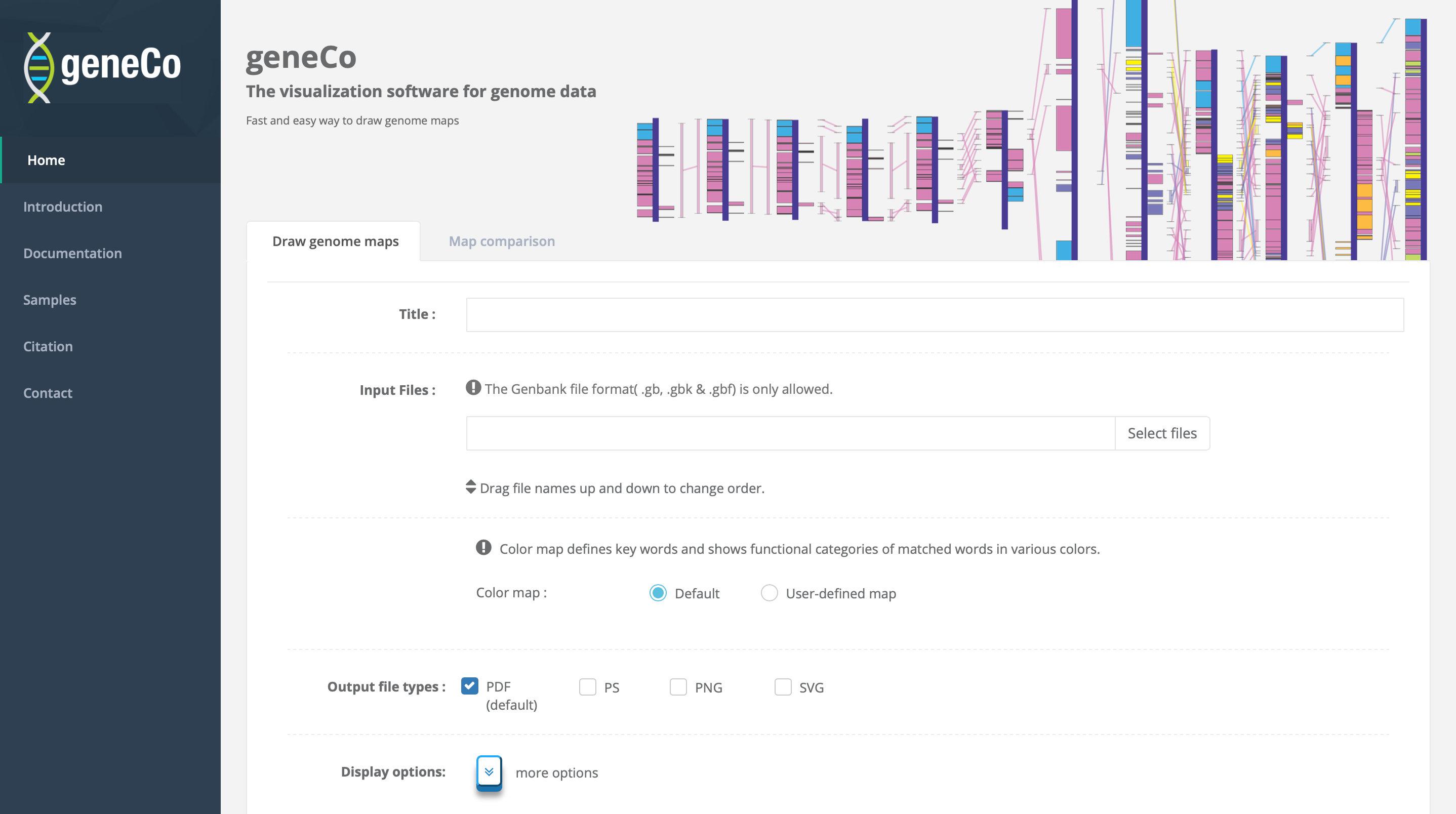
geneCo
geneCo: A visualized comparative genomic method to analyze multiple genome structures method to analyze multiple genome structures In comparative and evolutionary genomics, a detailed comparison of common features between organisms...
2019-01-27
Recruiting researchers
Looking for passionate Ph. D. and M.S. students who want to study the research areaa of data science, computational biology and bioinformatics.
데이터 사이언스 및 바이오인포매틱스 연구 분야를 공부하고자 하는 박사/석사 과정 학생을 찾습니다.